
Media
@macromoltek
Sept. 23, 2019
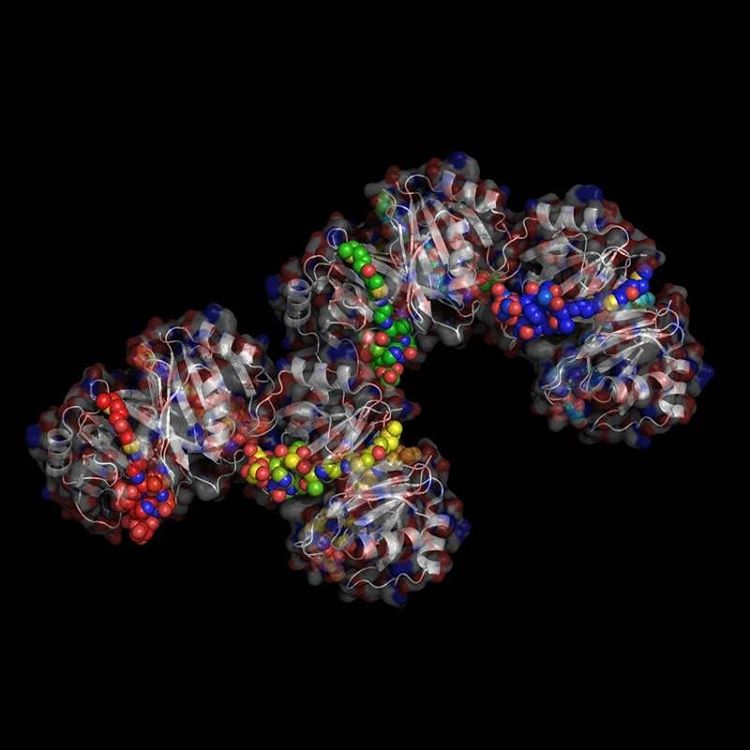
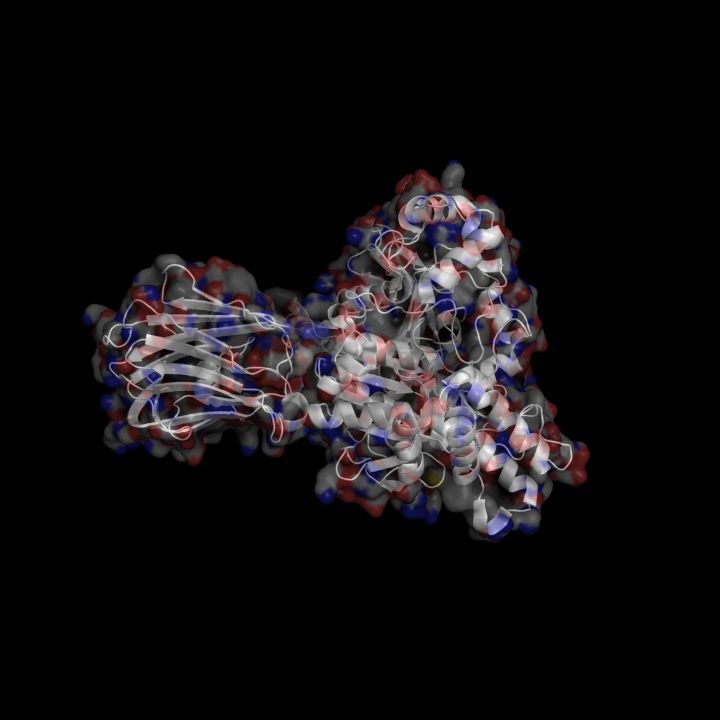
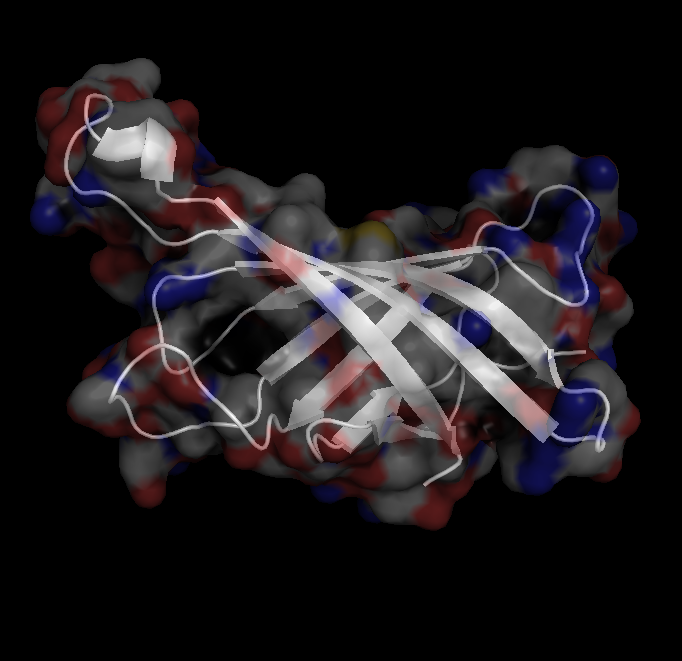
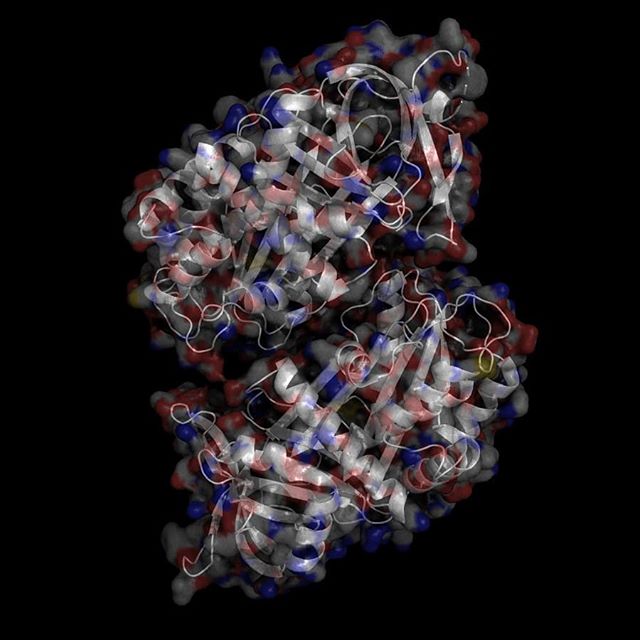
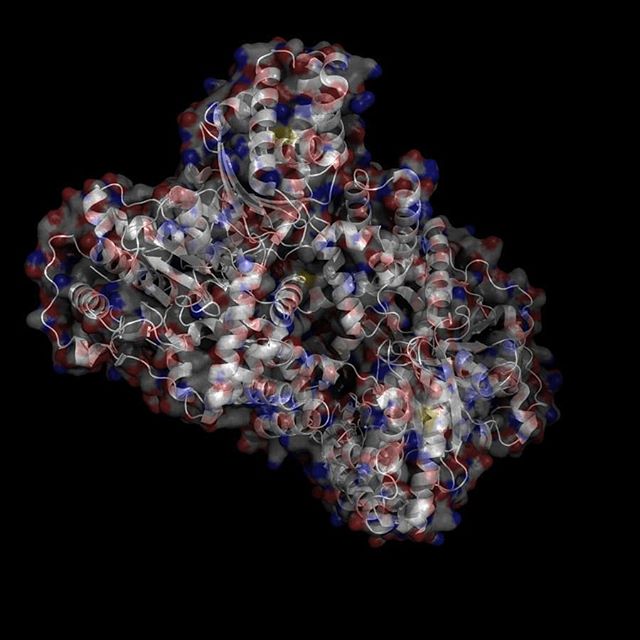
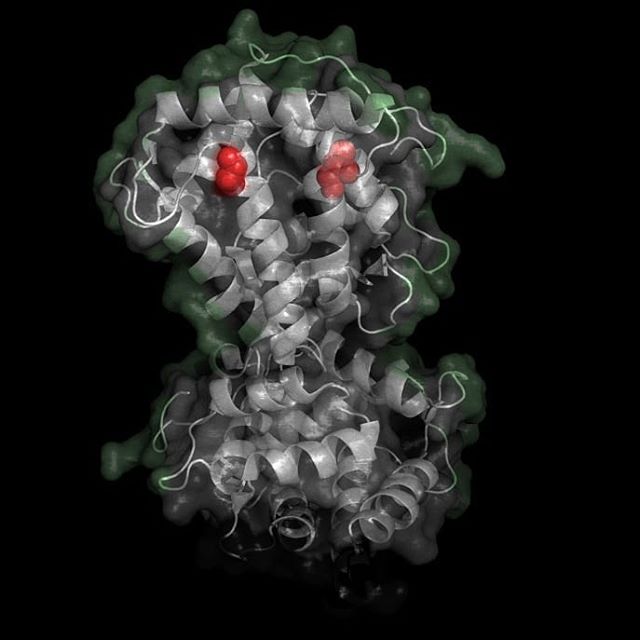
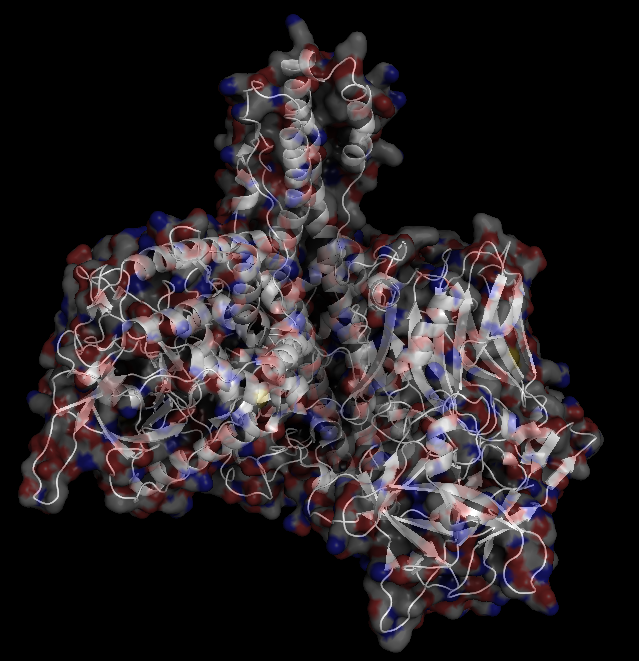
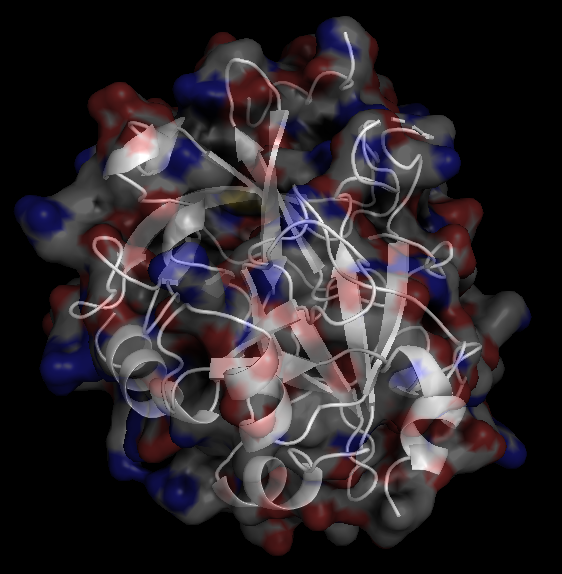
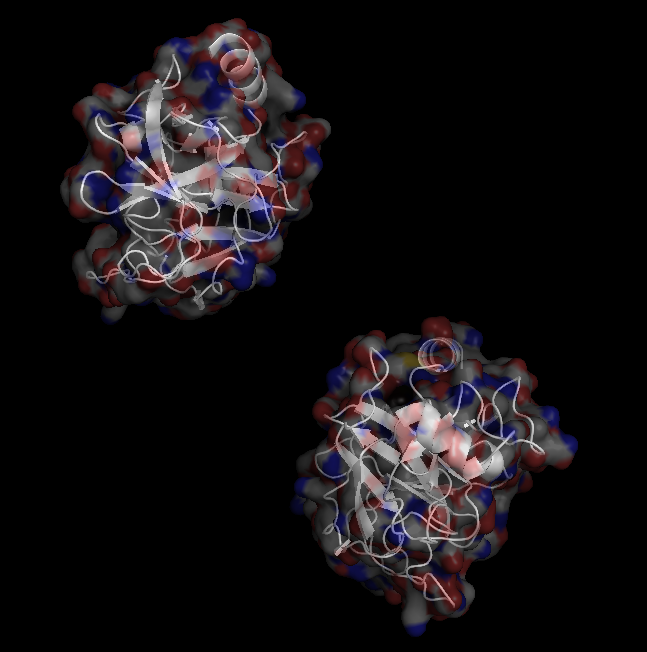
X-Ray Crystallography is no easy process, and it has its limitations as we've previously described in one of our blog posts [http://ow.ly/qN0150w4fQJ]. A big challenge stems from the inconsistencies during phasing resulting from X-ray absorption artifacts that lead to inconsistent sampling of crystals. One of the ways scientists have attempted to solve this issue is by strategically processing the proteins as they experience radiation damage and then relying on powerful software to identify the resulting defects and revert the changes. Shown on the top left is a radiated sample of Bovine Trypsin (PDB ID: 1n6y). This is one of the first protein structures to be tested using the technique known as Radiation-damage Induced Phasing method (RIP). The image on the bottom right is for comparison. This is PDB ID 1s0q which is Bovine Trypsin imaged using x-ray crystallography.
All Rights Reserved